Синонимы: ПКВКЛ, pCBCL
Главное
- Кожные B-клеточные лимфомы представляют собой группу лимфом, первичным очагом которых является кожа; они происходят из B-лимфоцитов на разных стадиях дифференцировки. Кожа также может быть местом вторичного поражения при внекожных (обычно нодальных) B-клеточных лимфомах.
- Большинство первичных кожных B-клеточных лимфом (>80%) относятся к опухолям низкой степени злокачественности и характеризуются индолентным течением и благоприятным прогнозом.
- Точная классификация возможна только после комплексной оценки клинических, гистопатологических, иммунофенотипических и молекулярных признаков. Классификации WHO-EORTC и ICC служат основой для последовательной и унифицированной классификации пациентов.
- Варианты лечения низкозлокачественных форм включают прежде всего выжидательную тактику, внутриочаговое введение кортикостероидов и локальную лучевую терапию; при наличии солитарных очагов возможна хирургическая эксцизия. У части пациентов могут применяться системная или внутриочаговая терапия анти-CD20-антителами, тогда как системная химиотерапия обычно требуется только пациентам с высокозлокачественными опухолями.
Введение
Хотя B-клеточные лимфомы составляют большинство неходжкинских лимфом (НХЛ), возникающих в лимфатических узлах, среди НХЛ с первичным поражением кожи они встречаются реже. В классификации кожных лимфом, опубликованной Всемирной организацией здравоохранения (WHO) и исследовательской группой по кожным лимфомам Европейской организации по исследованию и лечению рака (EORTC), B-клеточные лимфомы составляли около 25% всех кожных лимфом. Классификации WHO-EORTC 2005 и 2018 годов были интегрированы с минимальными изменениями в International Consensus Classification (ICC) зрелых лимфоидных неоплазий, опубликованную в 2022 году.
Первичные кожные лимфомы определяются как: злокачественные лимфомы, ограниченные кожей на момент установления диагноза после полного стадирования.
В целом большинство пациентов с первичной кожной B-клеточной лимфомой (ПКВКЛ) впервые диагностируются дерматологами, поскольку внекожные симптомы и признаки в дебюте заболевания наблюдаются крайне редко. Поэтому дерматологам необходимо хорошо ориентироваться в клинико-патоморфологических особенностях этой группы заболеваний, чтобы иметь возможность устанавливать диагноз на ранних стадиях. Кроме того, поскольку агрессивные методы лечения требуются лишь в отдельных случаях, ведение таких пациентов должно преимущественно осуществляться дерматологами, имеющими специальную подготовку в области кожных лимфом.
Историческая справка
- В прошлом считалось, что кожные B-клеточные лимфомы всегда являются вторичными, то есть возникают вследствие диссеминации внекожных, обычно нодальных, B-клеточных лимфом в кожу.
- В 1980-х годах было установлено, что B-клеточные лимфомы, первичным очагом которых является кожа, то есть ПКВКЛ, представляют собой самостоятельную группу экстранодальных лимфом.
- Широкое внедрение иммуногистохимических и молекулярно-генетических методов, позволяющих установить клональность, в частности выявление перестроек генов иммуноглобулинов методом ПЦР с использованием рутинных биопсийных образцов, показало, что часть случаев, ранее классифицировавшихся как кожные B-клеточные псевдолимфомы, содержит моноклональную популяцию лимфоцитов и, следовательно, на самом деле представляет собой низкозлокачественные B-клеточные лимфомы кожи.
Эпидемиология
В течение последних нескольких десятилетий заболеваемость первичными кожными В-клеточными лимфомами росла, однако в настоящее время, по-видимому, стабилизировалась. ПКВКЛ могут встречаться чаще в определённых регионах мира. Например, анализ данных четырёх академических центров США показал, что они составляли лишь 4,5% всех случаев первичных кожных лимфом, зарегистрированных в этих центрах. Напротив, в нидерландском и австрийском регистрах кожных лимфом ПКВКЛ составляли около 25% всех случаев первичных кожных лимфом.
Региональные различия отмечались и в отношении частоты отдельных типов ПКВКЛ. В двух крупнейших опубликованных сериях наблюдений относительная частота лимфомы из клеток фолликулярного центра и лимфопролиферативного заболевания из клеток маргинальной зоны значительно различалась. Лимфома из клеток фолликулярного центра составляла 71% всех ПКВКЛ в Нидерландах, но только 41% в Граце; напротив, лимфопролиферативное заболевание из клеток маргинальной зоны (которое в то время классифицировалось как лимфома) составляло 42% всех случаев ПКВКЛ в Граце, но лишь 10% в Нидерландах. В то же время относительная частота третьего по распространённости типа ПКВКЛ — диффузной крупноклеточной В-клеточной лимфомы, leg type — была сходной в обеих сериях (около 15%), что указывает на то, что различия в относительной частоте в этих двух сериях касались только В-клеточных лимфом с индолентным течением. Это позволяет предположить, что такие различия могут быть, по крайней мере частично, обусловлены различиями в классификации пациентов в разных центрах. 1
Помимо различий в диагностических и классификационных подходах, истинные региональные различия в частоте отдельных типов ПКВКЛ могут быть связаны с наличием различных этиологических факторов. Например, связь между ПКВКЛ и инфекцией, вызванной определёнными подвидами Borrelia, в эндемичных районах известна уже много лет. Эта связь может частично объяснять региональные различия в частоте ПКВКЛ, однако относительно небольшая доля таких случаев даже в странах, эндемичных по Borrelia, позволяет предполагать участие и других этиологических факторов.
ПКВКЛ поражают взрослых обоих полов. За исключением лимфобластной В-клеточной лимфомы/лейкоза из предшественников В-клеток, который лишь в редких случаях действительно представляет собой пример первичной кожной В-клеточной лимфомы, кожные В-клеточные лимфомы редко встречаются у детей и подростков.
Этиология и патогенез
Патогенез ПКВКЛ неизвестен. В отличие от многих нодальных В-клеточных лимфом, связанных со специфическими генетическими нарушениями, например нодальных фолликулярных лимфом с межхромосомной транслокацией 14;18, данные о специфических генетических особенностях ПКВКЛ, особенно их низкозлокачественных подтипов, остаются ограниченными.
Некоторое сходство с клинико-патоморфологическими признаками В-клеточных лимфом, возникающих в слизистой оболочке желудка, так называемых лимфом, ассоциированных со слизистой оболочкой (MALT-лимфом), привело к гипотезе о том, что ПКВКЛ обусловлены длительной антигенной стимуляцией, возможно связанной с хронической инфекцией определёнными микроорганизмами. Действительно, давно установлено, что MALT-лимфомы желудка связаны с хронической антигенной стимуляцией вследствие инфекции Helicobacter pylori. Как уже отмечалось, Borrelia spp., предположительно, играют этиологическую роль в меньшинстве случаев ПКВКЛ, особенно в Европе. У пациентов из региона Австрии, эндемичного по боррелиозу, специфические последовательности ДНК Borrelia были выявлены в 18% всех случаев ПКВКЛ; сходные данные были получены и в Шотландии.
На сегодняшний день ни один другой микроорганизм не был убедительно связан с развитием ПКВКЛ. Однако следует отметить, что ПКВКЛ описаны у пациентов с иммуносупрессией, включая больных СПИДом и реципиентов солидных органов, а также наблюдались обратимые формы ПКВКЛ у пациентов, получавших терапию метотрексатом, особенно при ревматоидном артрите. Это позволяет предполагать, что иммунная дисрегуляция может играть роль в развитии данного заболевания.
Различные формы B-клеточных лимфом
Классифакация
В классификациях WHO-EORTC 2018 и ICC 2022 ПКВКЛ подразделяются на пять основных типов + неуточненный вариант.
| WHO-EORTC 2018 | ICC 2022 |
|---|---|
| • Первичная кожная лимфома из клеток фолликулярного центра | • Первичная кожная лимфома из клеток фолликулярного центра |
| • Первичная кожная B-клеточная лимфома маргинальной зоны* | • Первичное кожное B-клеточное лимфопролиферативное заболевание маргинальной зоны |
| • Первичная кожная диффузная крупноклеточная B-клеточная лимфома, leg type | • Первичная кожная диффузная крупноклеточная B-клеточная лимфома, leg type |
| • Первичная кожная диффузная крупноклеточная B-клеточная лимфома, другой тип | • Диффузная крупноклеточная B-клеточная лимфома, NOS |
| • EBV+ слизисто-кожная язва (предварительная категория) | • ЭБВ+ слизисто-кожная язва |
| • Внутрисосудистая диффузная крупноклеточная B-клеточная лимфома | • Внутрисосудистая диффузная крупноклеточная B-клеточная лимфома |
NOS - not otherwise specified, неуточненный вариант
ВЭБ - вирус Эпштейна-Барр
* Включает случаи, ранее обозначавшиеся как первичная кожная иммунцитома и первичная кожная плазмоцитома.
ICC — International Consensus Classification
Лимфома из клеток фолликулярного центра и В-клеточное лимфопролиферативное заболевание маргинальной зоны характеризуются индолентным клиническим течением, тогда как диффузная крупноклеточная В-клеточная лимфома (leg type) и внутрисосудистая диффузная крупноклеточная В-клеточная лимфома отличаются агрессивным клиническим течением. Ещё один тип, Эпштейна–Барр-вирус-позитивная слизисто-кожная язва, также характеризуется индолентным течением.
Следует подчеркнуть, что, помимо ПКВКЛ, кожа может быть местом вторичного поражения практически при всех типах внекожных, обычно нодальных, В-клеточных лимфом и лейкозов. Всем пациентам с подтверждённым диагнозом кожного поражения при высокозлокачественной В-клеточной лимфоме необходимо полное стадирование. Оно включает общий анализ крови, проточную цитометрию периферической крови, КТ органов грудной клетки, брюшной полости и таза, а при возможности — позитронно-эмиссионную томографию (ПЭТ).
С другой стороны, при кожных низкозлокачественных В-клеточных лимфомах без внекожной симптоматики КТ органов грудной клетки, брюшной полости и таза, функциональная визуализация (ПЭТ-КТ) или биопсия костного мозга лишь в редких случаях приводят к изменению стадии заболевания в сторону её повышения. Поэтому расширенное лучевое обследование и биопсия костного мозга считаются избыточными при стадировании и последующем наблюдении пациентов с индолентными типами ПКВКЛ, ограниченными кожей.
Рекомендуемый объём стадирующего обследования у пациентов с кожными В-клеточными лимфомами:
| Исследование | Рекомендуется |
|---|---|
| Анамнез и физикальное обследование | |
| Наличие/отсутствие B-симптомов, включая лихорадку, ночную потливость, снижение массы тела, недомогание | √ |
| Полное обследование лимфатических узлов | √ |
| Пальпация живота для выявления гепатоспленомегалии | √ |
| Осмотр полости рта | √ |
| Анамнез трансплантации органов и/или другой иммуносупрессии (например, ВИЧ, ятрогенной, врождённой) | √ |
| Лабораторные исследования | |
| Общий анализ крови с лейкоцитарной формулой и подсчётом тромбоцитов | √ |
| Развёрнутый биохимический анализ крови, включая ЛДГ | √ |
| Проточная цитометрия мононуклеаров периферической крови | √° |
| Методы визуализации | |
| УЗИ брюшной полости и поверхностных лимфатических узлов как скрининг перед ПЭТ-КТ или КТ | √ |
| ПЭТ-КТ всего тела или КТ, если ПЭТ-КТ недоступна | √* |
| Дополнительные исследования по клиническим показаниям | |
| Биопсия костного мозга | √*,† |
| Эксцизионная биопсия увеличенных лимфатических узлов или других подозрительных очагов | √* |
| ПЦР-исследование ДНК Borrelia spp. у пациентов из эндемичных районов (Европа) или посещавших эндемичные районы | √ |
° При низкой степени злокачественности без внекожных симптомов — опционально.
* При низкой степени злокачественности без внекожных симптомов — только при подозрении на внекожную лимфому.
† В США биопсия костного мозга нередко заменяется проточной цитометрией в сочетании с ПЭТ-КТ.
ЛДГ — лактатдегидрогеназа; ВИЧ — вирус иммунодефицита человека; ПЦР — полимеразная цепная реакция; КТ — компьютерная томография; ПЭТ-КТ — позитронно-эмиссионная томография, совмещённая с компьютерной томографией.
Таблица дифференциальной диагностики
| Критерий | Первичная кожная лимфома из фолликулярного центра (PCFCL) | Вторичная фолликулярная лимфома | Первичная кожная лимфома маргинальной зоны | Первичная кожная диффузная крупноклеточная B-клеточная лимфома, тип нижних конечностей |
|---|---|---|---|---|
| Возраст | 50–60 лет | Около 60 лет | Около 55 лет | 70 лет |
| Пол | М > Ж | М > Ж | М > Ж | Ж > М |
| Стадия | Низкая (I–II) | В большинстве случаев высокая (III–IV) | Низкая, ограничена кожей | От низкой до высокой |
| Наиболее частая локализация | Голова и шея | Вариабельная | Туловище, верхние конечности или голова | Нижние конечности |
| Экстракутанное распространение | Отсутствует | Присутствует, вариабельно | Редко (<10%) | Присутствует, около 30% |
| Гистологическая картина | Смесь центроцитов и центробластов; при фолликулярном, фолликулярно-диффузном и диффузном типах роста градация не применяется; характерны фоновый фиброз и склероз со стромальной реакцией | Смесь центроцитов и центробластов в фолликулах; проводится градация по числу центробластов; характерны фоновый фиброз и склероз со стромальной реакцией | Полиморфный инфильтрат: мелкие лимфоциты, мелкие и средние клетки маргинальной зоны (центроцитоподобные) с бледной цитоплазмой, лимфоплазмоцитарные клетки, плазматические клетки и иногда крупные трансформированные лимфоидные клетки | Центробласты или иммунобласты (крупные клетки с нерасщеплёнными ядрами) при диффузном типе роста; стромальная реакция фона отсутствует |
| BCL2 (ИГХ) | Отрицательная или слабоположительная экспрессия | Обычно положительная экспрессия | Обычно положительная экспрессия | Положительная экспрессия |
| CD10 | Положительная экспрессия при фолликулярном паттерне (<25% случаев); при диффузном паттерне отрицательная | Обычно положительная экспрессия | Отрицательная экспрессия | Отрицательная экспрессия |
| BCL6 (ИГХ) | Положительная экспрессия (~100%) | Обычно положительная экспрессия | Отрицательная экспрессия | Экспрессия вариабельна: положительная или отрицательная |
| Ki-67 | Вариабелен, обычно >30% | Низкий, за исключением high-grade вариантов | — | — |
| Реаранжировка BCL2 / t(14;18) | Обычно отсутствует, однако в 10–40% случаев может выявляться | Положительная примерно в 85% случаев | Отсутствует | Выявляется примерно в 50% случаев |
| Реаранжировки BCL6 или MYC | Отсутствуют | Выявляются вариабельно | Редкие случаи с реаранжировкой BCL6 | BCL6 — около 30%, MYC — около 30% |
| Цитогенетические находки | Делеция 14q32.33; амплификация c-REL (~60%) | Комплексные изменения | • t(14;18)(q32;q21); IGH-MALT1 • t(11;18)(q21;q21); BIRC3-MALT1 • t(3;14)(p14.1;q32); IGH-FOXP1 | Инактивация или делеция CDKN2A (9p21.3) и CDKN2B; делеция 6q-плеча (BLIMP1; 60%) |
| Молекулярные находки | Мутации в генах ремоделирования хроматина (CREBBP, EZH2, KMT2D, EP300) встречаются менее чем в 10% случаев; профиль экспрессии генов сходен с GCB-подтипом ДВККЛ | Часто выявляются мутации в генах ремоделирования хроматина (CREBBP, EZH2, KMT2D, EP300) | MYD88 (6%), FAS (63%), SLAMF1 (24%), SPEN (18%) и NCOR2 (13%) | MYD88 (60%), CD79B (20%), CARD11 (10%), TNFAIP3/A20 (40%); профиль экспрессии генов сходен с ABC-подтипом ДВККЛ |
| Прогноз | Благоприятный; 5-летняя безрецидивная выживаемость около 95% | Вариабельный; 5-летняя выживаемость 20–75% | Очень благоприятный; 5-летняя выживаемость 95–100% | Вариабельный; 5-летняя выживаемость около 50% |
Источники: https://pubmed.ncbi.nlm.nih.gov/33560380/, https://pubmed.ncbi.nlm.nih.gov/32528871/, https://pubmed.ncbi.nlm.nih.gov/32808967/
Первичная кожная лимфома из клеток фолликулярного центра (ПКЛФЦ)
Первичная кожная лимфома из клеток фолликулярного центра (ПКЛФЦ) определяется как неопластическая пролиферация клеток герминативного центра, ограниченная кожей. Это один из частых подтипов первичных кожных B-клеточных лимфом.
Клинически у пациентов выявляются солитарные или сгруппированные папулы, бляшки или опухолевые узлы розового, багрово-розового или сливового оттенка, которые, особенно на туловище, могут быть окружены участками эритемы (см. фото ниже). Термин «лимфома Крости» использовали для обозначения случаев, при которых основной очаг был окружён периферическим эритематозным венчиком. Изъязвление встречается редко. Иногда заболевание проявляется милиарными и/или сгруппированными мелкими папулами, которые могут иметь акнеформный вид. Наиболее типичная локализация — волосистая часть головы, лоб и спина. Кожные высыпания обычно бессимптомны, а B-симптомы, то есть лихорадка, ночная потливость и снижение массы тела, как правило, редки. Уровень лактатдегидрогеназы (ЛДГ) в сыворотке, являющийся важным прогностическим фактором при системных лимфомах, обычно находится в пределах нормы.
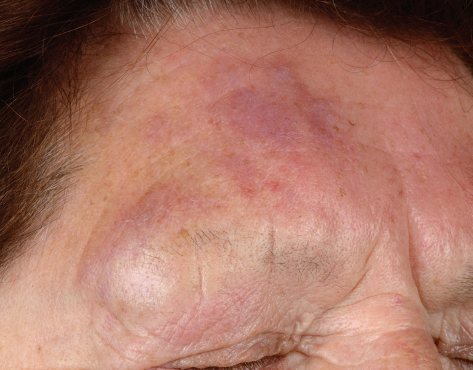
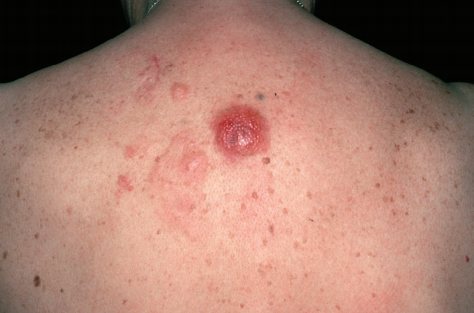
Прогноз благоприятный. Рецидивы наблюдаются почти у 50% пациентов, однако распространение в лимфатические узлы или внутренние органы происходит редко.
Патоморфология
Как оценивать лимфоцитарный инфильтрат?
- При ПКЛФЦ в толще всей дермы выявляются узловые или диффузные инфильтраты, нередко распространяющиеся в подкожную жировую клетчатку.
- Эпидермис обычно не вовлекается.
- Выделяют 3 варианта архитенктоники:
- фолликулярный
- фолликулярно-диффузный
- диффузный
Чёткая фолликулярная архитектура (фолликулы как в лимфатических узлах) с формированием неопластических герминативных центров выявляется у меньшинства пациентов (25%). Однако у большего числа случаев могут определяться хотя бы отдельные элементы фолликулярной архитектуры. В случаях с фолликулярным строением неопластические фолликулы имеют морфологические признаки злокачественности, включая истончение или отсутствие мантийной зоны, отсутствие макрофагов с «окрашивающимися тельцами» (tingible body macrophages), а также мономорфизм фолликулов — так называемые «тёмные» и «светлые» зоны более не различимы.
Как при фолликулярном, так и при диффузном варианте в составе неопластического инфильтрата преобладают центроциты — малые или крупные клетки фолликулярного центра с расщеплёнными ядрами; в различном количестве с ними сочетаются центробласты — крупные нерасщеплённые клетки фолликулярного центра с заметными ядрышками, иммунобласты, мелкие лимфоциты, гистиоциты, а в некоторых случаях - эозинофилы и плазматические клетки.
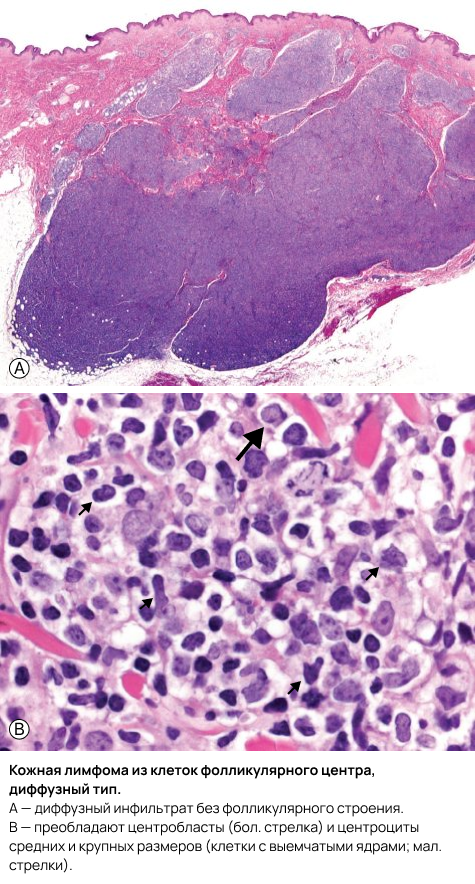
В отличие от нодальной фолликулярной лимфомы, градация по степени злокачественности при классификации ПКЛФЦ не используется. На цитоморфологическом уровне некоторые случаи ПКЛФЦ с диффузным типом роста и преобладанием крупных центроцитов могут напоминать крупноклеточную лимфому, однако их прогноз сопоставим с прогнозом очагов без крупноклеточной морфологии. Случаи с монотонной пролиферацией центробластов и/или иммунобластов следует классифицировать как диффузную крупноклеточную B-клеточную лимфому. Обычно также выявляется сопутствующий инфильтрат из мелких T-лимфоцитов и гистиоцитов/макрофагов; в отдельных случаях именно эти клетки могут преобладать. В редких наблюдениях может обнаруживаться популяция моноклональных плазматических клеток, принадлежащих неопластическому клону, что представляет собой диагностическую ловушку при дифференциальной диагностике с ПКЛПЗМЗ.
Помимо случаев с большим количеством крупных клеток, к морфологическим вариантам ПКЛФЦ относятся формы с преобладанием веретеноклеточной морфологии, которые гистопатологически могут имитировать саркомы или другие веретеноклеточные опухоли и тем самым создавать диагностические трудности. Поражения, в которых немногочисленные B-клеточные бласты сочетаются с большим количеством T-лимфоцитов, были отнесены к «богатым T-клетками/гистиоцитами B-клеточным лимфомам»; в коже они, вероятно, представляют ещё один редкий морфологический вариант ПКЛФЦ. Следует отметить, что при поражении лимфатических узлов этот последний морфологический вариант классифицируется как крупноклеточная B-клеточная лимфома.
Опухолевые клетки экспрессируют B-клеточные антигены (CD20, CD79a, PAX5); они CD5- и CD43-. При наличии фолликулов для них характерна неправильная сеть CD21+ фолликулярных дендритных клеток. Bcl-6, маркёр клеток герминативного центра и некоторых других лимфоидных клеток, положителен практически во всех случаях независимо от характера роста. В большинстве случаев с фолликулярным типом роста и у меньшинства случаев с диффузным типом роста неопластические клетки также позитивны по CD10. Наличие небольших скоплений Bcl-6+ клеток вне фолликулов считается признаком, убедительно указывающим в пользу диагноза ПКЛФЦ. Следует учитывать, что Bcl-6 экспрессируется и частью T-лимфоцитов — фолликулярными T-хелперами, однако при ПКЛФЦ эти клетки никогда не образуют скоплений.
В подавляющем большинстве случаев окрашивание на белковый продукт гена BCL2 (Bcl-2) в неопластических фолликулах даёт отрицательный результат, что представляет собой важное отличие от фолликулярных лимфом лимфатических узлов. Позитивность по Bcl-2 более чем в 75% неопластических лимфоцитов была связана с более высокой частотой кожных рецидивов. Полезным, хотя и несколько парадоксальным, иммуногистохимическим признаком является более низкая пролиферативная активность в пределах злокачественных фолликулов по данным окрашивания антителом к Ki-67, в отличие от выраженной Ki-67-позитивности в реактивных фолликулах. В случаях с крупноклеточной морфологией, то есть при преобладании крупных клеток с расщеплёнными ядрами, экспрессия MUM1 либо отсутствует, либо определяется лишь в небольшом числе опухолевых клеток, в отличие от выраженной экспрессии MUM1 при ПКДКВКЛ-ТНК. Межхромосомная транслокация t(14;18), типичная для нодальных фолликулярных лимфом, выявляется лишь у меньшинства случаев ПКЛФЦ. Поэтому наличие транслокации t(14;18) и/или экспрессии Bcl-2 должно вызывать подозрение на системную лимфому с поражением кожи. Анализ генов, кодирующих JH-сегмент и другие участки тяжёлой цепи иммуноглобулина (IGH), выявляет моноклональную перестройку у большинства пациентов (60–70%). При исследовании экспрессии генов в ПКЛФЦ с помощью кДНК-микрочипов был выявлен сигнатурный паттерн клеток герминативного центра. Совокупный анализ морфологических, иммуногистохимических и молекулярных данных позволяет предположить, что фолликулярные лимфомы, возникающие в лимфатических узлах и в коже, несмотря на сходный морфологический рисунок, имеют разные патогенетические механизмы.
Первичное кожное B-клеточное лимфопролиферативное заболевание маргинальной зоны
Первичное кожное B-клеточное лимфопролиферативное заболевание маргинальной зоны (ПКЛПЗМЗ) признано самостоятельным вариантом низкозлокачественной первичной кожной B-клеточной лимфомы. В классификации ICC 2022 оно больше не объединяется с другими лимфомами маргинальной зоны, ассоциированными со слизистой оболочкой, и понижено в статусе до лимфопролиферативного заболевания, а не истинной лимфомы. В 5-м издании классификации WHO оно по-прежнему приводится как лимфома. Следует отметить, что случаи, ранее классифицировавшиеся как первичная кожная иммунцитома или первичная кожная плазмоцитома, представляют собой варианты ПКЛПЗМЗ с выраженной лимфоплазмоцитарной или плазмоцитарной дифференцировкой соответственно, а термины «кожная иммунцитома» и «кожная плазмоцитома» в классификациях WHO-EORTC и ICC 2022 не используются.
Клинически у пациентов наблюдаются рецидивирующие папулы, бляшки и узлы розово-фиолетового, красно-коричневого цвета, преимущественно на конечностях, чаще на верхних, чем на нижних, либо на туловище (см. фото ниже). У небольшого числа пациентов поражение может быть генерализованным. Изъязвление встречается редко, и кожные высыпания обычно бессимптомны. Как правило, B-симптомы, то есть лихорадка, ночная потливость и снижение массы тела, отсутствуют. Уровень ЛДГ в сыворотке остается в пределах нормы. В ряде случаев разрешение очагов может сопровождаться вторичной анетодермией вследствие утраты эластических волокон в зоне опухолевого инфильтрата.
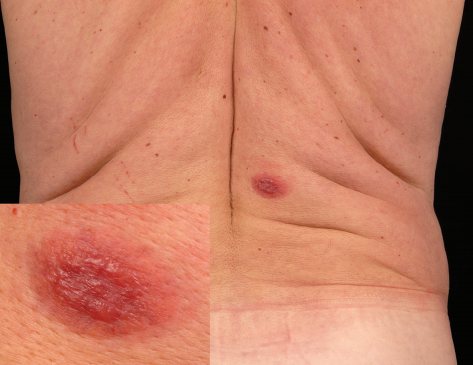
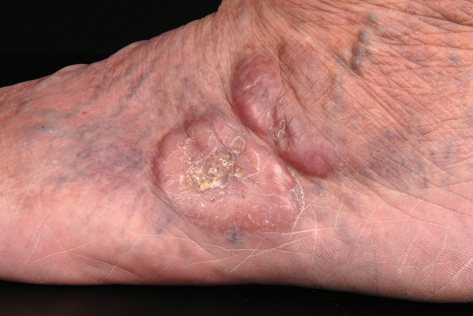
Прогноз при ПКЛПЗМЗ отличный. В исследовании 32 пациентов с ПКЛПЗМЗ ни у одного из них не развилось поражение лимфатических узлов или внутренних органов при среднем периоде наблюдения более 4 лет.
Следует также отметить, что ПКЛПЗМЗ может возникать в зонах, поражённых хроническим атрофическим акродерматитом, и, по-видимому, чаще, чем другие типы первичных кожных B-клеточных лимфом, быть связано с инфекцией Borrelia spp., особенно в Европе.
Патоморфология
- Гистологически для ПКЛПЗМЗ характерен пятнистый, узловой или диффузный инфильтрат, вовлекающий дерму и подкожную жировую клетчатку. Эпидермис при этом не поражается.
- При малом увеличении можно наблюдать характерную картину: узловые инфильтраты, иногда содержащие реактивные герминативные центры, окружены бледно окрашенной популяцией клеток малого и среднего размера с изрезанными ядрами, незаметными ядрышками и обильной светлой цитоплазмой. Эти клетки в разных источниках описываются как клетки маргинальной зоны, centrocyte-like cells или monocytoid B cells. Кроме того, в инфильтрате обнаруживаются плазматические клетки, преимущественно по его периферии, лимфоплазмоцитоидные клетки, мелкие лимфоциты и отдельные крупные бластные клетки. Частой находкой являются также эозинофилы. У части пациентов может отмечаться гранулематозная реакция с эпителиоидными и гигантскими клетками. Случаи с преобладанием лимфоплазмоцитоидных лимфоцитов ранее относили к кожным иммуноцитомам, однако в настоящее время их рассматривают как вариант ПКЛПЗМЗ. Иногда выявляются PAS-положительные внутриядерные включения, тельца Датчера, которые представляют собой ценную диагностическую подсказку. В отдельных наблюдениях преобладающим клеточным компонентом являются плазматические клетки; хотя такие случаи раньше классифицировали как первичные кожные плазмоцитомы, в настоящее время их также рассматривают как вариант ПКЛПЗМЗ. Наконец, редкие случаи характеризуются преобладанием крупных бластоидных клеток, напоминающих плазмобласты. Солитарный кожный узловой амилоидоз (кожная амилоидома), содержащий небольшую моноклональную популяцию плазматических клеток, может представлять собой своеобразный редкий вариант ПКЛПЗМЗ.
Centrocyte-like клетки экспрессируют CD20, CD79a, Fc receptor-like 4 (FCRL4/IRTA1)26 и Bcl-2, но не экспрессируют CD5, CD10 и Bcl-6. В подавляющем большинстве случаев определяется внутрицитоплазматическая монотипическая экспрессия лёгких цепей иммуноглобулинов — либо κ, либо λ, но не обеих одновременно. Моноклональная популяция B-лимфоцитов нередко характерно располагается по периферии клеточных агрегатов. Окрашивание на маркеры пролиферации также показывает усиленную позитивность по периферии узелков, а также в реактивных герминативных центрах. Экспрессия IgG4 выявляется менее чем в половине случаев с плазмоцитарной дифференцировкой, однако не связана с системным IgG4-ассоциированным заболеванием. Моноклональная реаранжировка IGH обнаруживается в большинстве случаев, примерно в 60–80%.
ПКЛПЗМЗ экспрессируют иммуноглобулины после переключения классов; например, один и тот же клон может переключаться с IgM на IgG или IgA28. Хотя B-лимфоцит меняет продукцию антител одного класса на другой, он сохраняет аффинность к тому же антигену. Для ПКЛПЗМЗ также характерны аберрантные соматические гипeрмутации, позволяющие связывание с различными антигенами. Небольшая часть ПКЛПЗМЗ экспрессирует IgM, то есть относится к non-switched вариантам, однако это ассоциировано с более высокой частотой внекожной диссеминации.
В подгруппе ПКЛПЗМЗ, как и при MALT-лимфомах других локализаций, выявляется специфическая транслокация t(14;18)(q32;21), затрагивающая IGH и MALT1; она встречается примерно в 30% случаев. В исследованиях экспрессии генов с использованием кДНК-микрочипов для ПКЛПЗМЗ был показан профиль, соответствующий плазматическим клеткам. К другим генетическим нарушениям относятся трисомия 3, сопровождающаяся повышенной экспрессией FOXP1, и, редко, транслокации 11;18 или 3;14^30. Однако более чем у 50% пациентов никаких молекулярных нарушений не выявляется.
Первичная кожная диффузная крупноклеточная B-клеточная лимфома, leg type
Первичная кожная диффузная крупноклеточная B-клеточная лимфома, leg type (DLBCLLT), представляет собой форму первичной кожной B-клеточной лимфомы, которая характеризуется преобладанием крупных округлых клеток — центробластов и иммунобластов, позитивных по Bcl-2, MUM-1 (multiple myeloma oncogene 1, или interferon regulatory factor 4, экспрессируемый постгерминальными B-клетками и плазматическими клетками) и FOX-P1. Встречается почти исключительно у пожилых пациентов, преимущественно у женщин.
Клинически заболевание проявляется солитарными или сгруппированными эритематозными либо красно-коричневыми узлами, локализующимися главным образом на дистальных отделах одной ноги (см. фото). У некоторых пациентов очаги могут возникать на обеих нижних конечностях одновременно или с небольшим интервалом времени. Возможно также изъязвление. Рядом с более крупными узлами могут определяться мелкие эритематозные папулы. Следует подчеркнуть, что примерно у 20% пациентов опухоли со сходными морфологическими и фенотипическими признаками могут возникать и вне нижних конечностей, однако обозначение DLBCLLT при этом всё равно сохраняется.
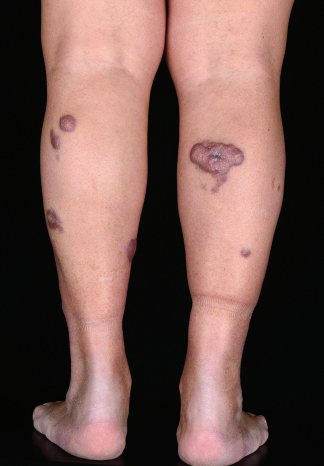
Прогноз при DLBCLLT менее благоприятный, чем при других типах первичных кожных B-клеточных лимфом: расчетная специфическая 5-летняя выживаемость составляет 50–60%.
Необходимо подчеркнуть, что у некоторых пациентов с нодальной крупноклеточной B-клеточной лимфомой вторичные кожные очаги могут локализоваться исключительно на ногах, что подтверждает необходимость полного стадирующего обследования до установления диагноза DLBCLLT. ПЭТ-исследование превосходит КТ в выявлении признаков системного заболевания.
Патоморфология
При DLBCL, leg type в дерме и подкожной клетчатке определяется плотный диффузный инфильтрат. Обычно он захватывает всю толщу сосочкового слоя дермы и доходит до дермо-эпидермального соединения. В некоторых случаях могут наблюдаться скопления крупных атипичных клеток в эпидермисе, имитирующие микроабсцессы Потрие, характерные для кожной T-клеточной лимфомы; это явление B-клеточного эпидермотропизма представляет потенциальную диагностическую ловушку.
Опухолевый инфильтрат состоит преимущественно из иммунобластов — крупных округлых клеток с обильной цитоплазмой и выраженными ядрышками — и центробластов. Следует отметить, что случаи первичных кожных B-клеточных лимфом с преобладанием крупных клеток с расщеплёнными ядрами, то есть крупных центроцитов, относят к первичной кожной фолликулярно-центровой лимфоме, а не к DLBCL, leg type. Реактивных мелких лимфоцитов обычно немного, митозы встречаются часто. Частое выявление гипермутаций генов иммуноглобулинов свидетельствует о том, что DLBCL, leg type представляет собой крупноклеточную лимфому из постгерминальных B-лимфоцитов, происходящих из клеток герминативного центра.
Опухолевые клетки экспрессируют B-клеточные маркеры, включая CD20, CD79a, PAX-5 и IgM, однако возможна частичная утрата экспрессии отдельных антигенов. В большинстве случаев опухолевые клетки также экспрессируют Bcl-2, MUM-1, FOX-P1 и MYC. Эти маркеры полезны для дифференциальной диагностики DLBCL, leg type и первичной кожной фолликулярно-центровой лимфомы диффузного типа: при последней Bcl-2, MUM-1 и FOX-P1 обычно либо отрицательны, либо экспрессируются лишь небольшим меньшинством клеток. В опухолях определяется моноклональная реаранжировка IGH. Межхромосомная транслокация t(14;18) отсутствует.
Редко в иных типичных случаях кожной DLBCL, leg type опухолевые клетки могут экспрессировать CD30. Для нодальных диффузных крупноклеточных B-клеточных лимфом экспрессия CD30 ассоциирована с более благоприятным прогнозом, однако для DLBCL, leg type таких данных пока недостаточно. У части пациентов наблюдается гиперметилирование генов p15 и/или p16 (CDKN2B, CDKN2A), что приводит к снижению экспрессии соответствующих белков. Делеции 9p21, потенциально обусловливающие loss-of-function CDKN2A, связаны с худшим прогнозом. Неблагоприятный прогноз также отмечен у пациентов с мутациями MYD88 в опухоли.
В нодальных диффузных крупноклеточных B-клеточных лимфомах перестройка MYC в сочетании с дополнительными перестройками BCL-2 и/или BCL-6, так называемые double-hit или triple-hit лимфомы, ассоциирована с худшим прогнозом. Неблагоприятный прогноз также связан с экспрессией соответствующих белков, выявляемой иммуногистохимически, то есть с паттернами double expression или triple expression. Данные по первичным кожным B-клеточным лимфомам пока фрагментарны, однако, по-видимому, перестройки MYC встречаются в них чаще, чем в B-клеточных лимфомах внекожных локализаций, тогда как double-hit варианты крайне редки и, вероятно, не влияют на прогноз.
Экспрессия MYC, Bcl-2 и Bcl-6 нередко встречается при первичных кожных лимфомах, однако прогностическое значение double expression, если оно вообще имеется, пока остаётся неясным. Хотя пороговые значения для оценки позитивности Bcl-2 и MYC в клетках пока не полностью унифицированы, в настоящее время общепринятыми считаются cut-off 50% для Bcl-2 и 40% для MYC.
Генетические данные, полученные с помощью FISH и технологий микрочипов, подтвердили, что DLBCL, leg type имеет чёткие молекулярные отличия от первичной кожной фолликулярно-центровой лимфомы диффузного типа, что подтверждает необходимость их раздельной классификации. Молекулярный профиль DLBCL, leg type сходен с таковым при нодальных диффузных крупноклеточных B-клеточных лимфомах и соответствует сигнатуре активированного B-лимфоцита.
Внутрисосудистая диффузная крупноклеточная B-клеточная лимфома
Внутрисосудистая диффузная крупноклеточная B-клеточная лимфома (IVDLBCL) — редкая злокачественная пролиферация крупных B-лимфоцитов в просвете кровеносных сосудов. У большинства пациентов опухоль имеет B-клеточный фенотип, однако описан и T-клеточный вариант. В отдельных случаях кожа может быть единственным поражённым органом, хотя чаще уже с самого начала имеется системное поражение, включая центральную нервную систему.
Клинически заболевание проявляется уплотнёнными эритематозными или фиолетовыми пятнами и бляшками, преимущественно на туловище и бёдрах, нередко с выраженными телеангиэктазиями. Клиническая картина не является типичной для кожной лимфомы и иногда может напоминать панникулит или сосудистые опухоли. Интересно, что у ряда пациентов IVDLBCL наблюдали в пределах очагов вишневых ангиом. Прогноз при IVDLBCL неблагоприятный, заболевание обычно протекает агрессивно.
Патоморфология
IVDLBCL характеризуется пролиферацией крупных атипичных лимфоцитов, заполняющих расширенные кровеносные сосуды в дерме и подкожной клетчатке. Клетки опухоли крупные, со скудной цитоплазмой и нередко с выраженными ядрышками. Они экспрессируют B-клеточные маркеры, а также Bcl-2, MUM-1 и FOX-P1.
Окрашивание антителами к эндотелиальным маркерам, например CD31 и CD34, позволяет подчеркнуть характерную внутрисосудистую локализацию опухолевых клеток. Молекулярный анализ выявляет моноклональную реаранжировку IGH. Генетический профиль сходен с таковым при диффузной крупноклеточной B-клеточной лимфоме activated B-cell type.
Лимфобластная лимфома/лейкоз из предшественников B-клеток
B-клеточная лимфобластная лимфома/лейкоз представляет собой злокачественную пролиферацию предшественников B-лимфоцитов. Сообщения о пациентах, у которых первичным очагом была кожа, редки. Однако важно подчеркнуть, что всех таких пациентов следует рассматривать как имеющих системное заболевание и лечить соответственно, даже при отсутствии документированного системного поражения на момент первичного обращения.
В отличие от других кожных B-клеточных лимфом, это заболевание имеет отчётливую предрасположенность к развитию у детей и молодых взрослых.
Клинически у пациентов выявляют солитарные крупные эритематозные опухолевые узлы, чаще всего локализующиеся в области головы и шеи. У пациентов с первичным поражением кожи очаги нередко бессимптомны и существуют в течение нескольких недель. При вторичном поражении кожи могут наблюдаться системные симптомы, например снижение массы тела, лихорадка, утомляемость и ночная потливость. Уровень ЛДГ в сыворотке часто повышен, что отражает агрессивный и нередко системный характер заболевания. Заболевание отличается высокой агрессивностью, а прогноз при отсутствии лечения неблагоприятный.
Патоморфология
Гистологически precursor B lymphoblastic lymphoma/leukemia характеризуется мономорфной пролиферацией клеток среднего размера со скудной цитоплазмой и округлыми или извилистыми ядрами с нежным хроматином. При малом увеличении часто определяется картина «звездного неба», обусловленная наличием макрофагов с включениями, то есть tingible body macrophages. Еще одной характерной особенностью является расположение опухолевых клеток по типу «мозаичной мостовой». Митозы и некротические клетки многочисленны.
Следует подчеркнуть, что только на основании гистологической картины невозможно надежно отличить лимфобластные лимфомы B-клеточного фенотипа от лимфобластных лимфом T-клеточной линии. Иммуногистохимически выявляется положительное окрашивание на TdT (терминальную дезоксинуклеотидилтрансферазу, экспрессируемую клетками-предшественниками T- и B-линии), PAX-5, CD10 и цитоплазматическую μ-цепь иммуноглобулинов, а в большинстве случаев также на CD20 и CD79a. Вариант pre-pre-B-cell является CD20-негативным и CD34-позитивным.
Молекулярные исследования обычно выявляют моноклональную реаранжировку IGH и поликлональный паттерн генов T-клеточного рецептора (TCR), однако возможны и отсутствие реаранжировки IGH, и моноклональная реаранжировка одновременно как TCR, так и IGH
Другие кожные B-клеточные лимфомы
Редкие случаи первичных кожных B-клеточных лимфом не укладываются в перечисленные выше подтипы. В классификации ICC 2022 такие пациенты распределяются по нескольким категориям, включая, помимо прочего, крупноклеточные B-клеточные лимфомы, развивающиеся на фоне иммуносупрессии, плазмобластную лимфому и диффузную крупноклеточную B-клеточную лимфому без дополнительного уточнения (NOS).
Плазмобластная лимфома — редкая лимфома, которая обычно возникает в полости рта у пациентов с выраженной иммуносупрессией, особенно связанной с ВИЧ-инфекцией, однако может наблюдаться и у иммунокомпетентных лиц. Она часто ассоциирована с инфекцией, вызванной вирусом Эпштейна–Барр.
ВЭБ-позитивная слизисто-кожная язва — B-клеточное новообразование низкой степени злокачественности, обычно возникающее в условиях иммунного старения или у иммунокомпрометированных пациентов. Чаще всего она проявляется язвами слизистой оболочки полости рта.
Патоморфология
EBV-позитивная мукокутанная язва характеризуется наличием атипичных клеток, экспрессирующих CD30 и PAX5, на фоне полиморфного клеточного окружения, что может имитировать картину лимфомы Ходжкина. EBV выявляется в атипичных клетках, однако позитивность отмечается также и в мелких лимфоцитах, что служит важным признаком для дифференциальной диагностики с истинной лимфомой Ходжкина, при которой EBV-позитивность обычно ограничена крупными атипичными клетками. Кроме того, клинические проявления этих двух состояний принципиально различаются.
Плазмобластная лимфома характеризуется пролиферацией плазмобластов с крупными эксцентрично расположенными ядрами, обильной цитоплазмой и выраженными ядрышками. Хотя опухолевые клетки могут напоминать лимфоидные, по иммунному фенотипу они соответствуют плазматическим клеткам. Они экспрессируют CD38, CD138 и MUM-1, но не экспрессируют CD20; также для них характерна монотипическая экспрессия лёгких цепей иммуноглобулинов. Гибридизация in situ на EBV (EBER-1) оказывается положительной в подавляющем большинстве случаев.