Амилоидоз — это не одно заболевание; данный термин используют для обозначения нескольких заболеваний, общим признаком которых является аномальное внеклеточное отложение амилоида — фибриллярного белкового материала — в тканях.
Отложения амилоида могут наблюдаться при широком спектре клинических состояний: от плазмоклеточных дискразий и болезни Альцгеймера до семейных полинейропатий и первичного кожного лихеноидного амилоидоза.
Сам амилоид не представляет собой единое химически однородное вещество; описано несколько его типов. Однако независимо от источника, патогенетических механизмов или основного заболевания амилоидный материал обладает рядом общих тинкториальных и физико-химических свойств, например конфигурацией перекрёстно-β-складчатого слоя.
Две клинические ситуации, в которых дерматолог с наибольшей вероятностью сталкивается с амилоидозами, включают:
- более распространённые первичные кожные формы амилоидоза;
- более редкий системный амилоидоз с кожными проявлениями.
Историческая справка
- В 1854 году Virchow ввёл термин «амилоид». Он полагал, что это вещество напоминает крахмал или целлюлозу, поскольку, подобно крахмалу, оно окрашивалось в синий цвет при обработке йодом с последующим добавлением разбавленной серной кислоты.
- В 1928 году Gutmann впервые описал пациента с клиническими признаками лихеноидного амилоидоза, а в 1930 году Freudenthal предложил термин «lichen amyloidosus».
Патогенез
Основным компонентом амилоида является фибриллярный белок; к минорным компонентам относятся амилоидный P-компонент, гликозаминогликаны и аполипопротеин E. Амилоидный P-компонент представляет собой гликопротеин, происходящий из сывороточного амилоидного P-белка (SAP), и обладает специфической кальций-зависимой способностью связываться с амилоидом. Идентифицировано более 40 различных форм амилоидных фибриллярных белков и их предшественников, включая: AL-амилоид, содержащий лёгкие цепи иммуноглобулинов; AA-амилоид, состоящий из белка острой фазы, синтезируемого печенью; Aβ-амилоид, обнаруживаемый в церебральных очагах при болезни Альцгеймера; и ATTR-амилоид, выявляемый при некоторых формах семейного амилоидоза (мутантный вариант) и старческого системного амилоидоза (дикого типа). Каждое из этих состояний связано со специфическим белком-предшественником; первоначально такие предшественники являются растворимыми белками, которые затем претерпевают изменения, приводящие к агрегации, полимеризации, формированию фибрилл и, в конечном итоге, к внеклеточному отложению нерастворимого амилоида в тканях.
Процесс, посредством которого происходит эта трансформация, различается при разных типах амилоидоза. При первичном системном амилоидозе замена аминокислот в определённых позициях вариабельной области лёгкой цепи иммуноглобулина потенциально дестабилизирует эти цепи и тем самым повышает вероятность их превращения в амилоидные фибриллы. Аналогичным образом показано, что мутации в гене транстиретина изменяют стабильность белка транстиретина и повышают его исходную, хотя и умеренную, амилоидогенность. Накопление этих относительно инертных амилоидных фибрилл в жизненно важных органах приводит к нарушению их функции.
Точный патогенез первичного кожного амилоидоза до настоящего времени полностью не установлен. В качестве возможных этиологических факторов рассматриваются длительное трение, генетическая предрасположенность, например OSMR и IL31RA, а также факторы окружающей среды. Белок-предшественник, вовлечённый в этот процесс, окончательно не охарактеризован; однако считается, что при макулярном и лихеноидном вариантах первичного кожного амилоидоза он имеет кератиноцитарное происхождение. Это предположение подтверждается ультраструктурными исследованиями, показавшими переходные формы между жизнеспособными кератиноцитами и амилоидом, а также положительными реакциями с моноклональными антителами против кератинов базального слоя. Согласно фибриллярной теории, тонофиламенты кератиноцитов подвергаются дегенерации и переходят в дерму, где, предположительно, модифицируются гистиоцитами и фибробластами в амилоидный материал. Альтернативная теория предполагает, что этот материал образуется на уровне дермоэпидермального соединения, а белки-предшественники секретируются базальными кератиноцитами. Эту гипотезу поддерживают ультраструктурные находки, а также выявление в амилоидных депозитах антигенов базальной мембраны, таких как коллаген IV типа и ламинин. Хотя эти кожные амилоидные отложения положительно окрашиваются антителами против человеческих IgG, IgM и IgA, считается, что такое окрашивание обусловлено неспецифической абсорбцией иммуноглобулинов, а не тем, что иммуноглобулины являются предполагаемым белком-предшественником. Также показано, что с первичными кожными амилоидными отложениями ассоциированы аполипопротеин E4, галектин-7 и актин; возможно, они синтезируются локально кератиноцитами. При первичном кожном амилоидозе описана нейропатия тонких волокон, которая может быть связана с сопутствующим зудом.
При узловом амилоидозе специфическое окрашивание антителами к кератину отсутствует; напротив, амилоидные отложения состоят из лёгких цепей иммуноглобулинов, что указывает на их плазмоклеточное происхождение, аналогично кожным проявлениям первичного системного амилоидоза. Таким образом, происхождение узлового амилоидоза существенно отличается от такового при макулярном и лихеноидном амилоидозе. Предполагается, что при узловом амилоидозе лёгкие цепи продуцируются локально в коже, тогда как при первичном системном амилоидозе с поражением кожи они поступают из системного кровотока.
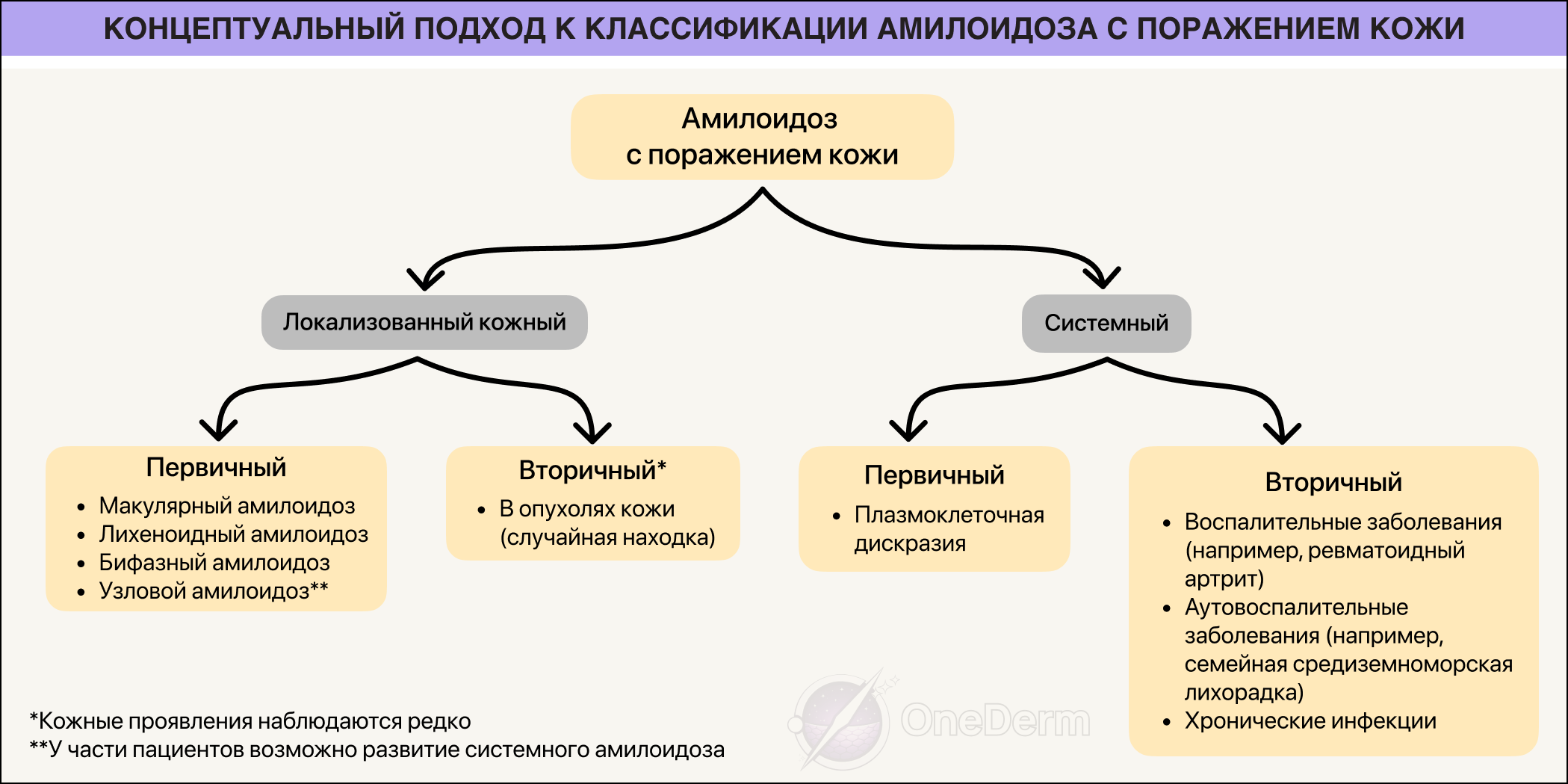
Клиническая классификация кожного амилоидоза
Первичный:
- макулярный,
- лихеноидный,
- бифазный,
- дисхромический,
- узловой
Вторичный:
- случайная находка в различных опухолях кожи (например, себорейный кератоз, базальноклеточный рак, дерматофиброма, внутридермальный меланоцитарный невус, опухоли придатков кожи, болезнь Боуэна, порокератоз);
- после ПУВА-терапии
Первичный локализованный кожный амилоидоз
Первичный кожный амилоидоз характеризуется отложением амилоида в коже без вовлечения внутренних органов.
Наиболее распространёнными вариантами являются:
- Макулярный амилоидоз,
- Лихеноидный амилоидоз,
- Бифазный амилоидоз.
Хотя первичный кожный амилоидоз традиционно подразделяют на макулярную форму (макулярный амилоидоз), папулёзную форму (лихеноидный амилоидоз) и узловую форму (узловой амилоидоз), первые две формы фактически представляют собой крайние точки единого клинического спектра. У части пациентов, а иногда и в пределах отдельных очагов, могут одновременно присутствовать как макулярные, так и папулёзные элементы; такую форму обозначают как «бифазный амилоидоз». Первичный кожный амилоидоз может существенно снижать качество жизни вследствие сопутствующего зуда и выраженного косметического дефекта вследствие кожных высыпаний.
Макулярный амилоидоз часто сопровождается зудом, однако в ряде случаев может протекать бессимптомно. Его элементы представлены гиперпигментированными очагами, имеющими либо сливной, либо рябящий рисунок. Последний лучше выявляется при натяжении кожи. Наиболее частая локализация — верхняя часть спины, особенно лопаточная область; реже поражаются разгибательные поверхности конечностей. Возможен линейный и даже невусоподобный рисунок очагов. Макулярный амилоидоз обычно дебютирует в молодом взрослом возрасте; женщины, возможно, страдают чаще мужчин. При бифазном амилоидозе на фоне гиперпигментации определяются мелкие папулёзные элементы.
Термин «фрикционный амилоидоз» используют для обозначения подгруппы пациентов, у которых локальное трение, обусловленное использованием нейлоновых щёток, полотенец и других грубых материалов, способствует формированию макулярных и лихеноидных очагов. Также существует значительное клиническое перекрытие между макулярным амилоидозом и пигментной формой notalgia paresthetica, при которой зуд в области лопаток приводит к растиранию и расчёсыванию кожи.
Лихеноидный амилоидоз является наиболее частой формой первичного кожного амилоидоза и обычно проявляется стойкими зудящими бляшками на передней поверхности голеней или других разгибательных поверхностях, например на передней поверхности бёдер или предплечьях. Начальные элементы представлены отдельными плотными шелушащимися папулами цвета кожи или гиперпигментированными папулами, которые впоследствии сливаются в бляшки, нередко приобретающие рябящий или гребешковидный рисунок. В начале заболевания очаги обычно односторонние, однако со временем может формироваться двустороннее симметричное распределение.
Описан также ано-сакральный вариант первичного кожного амилоидоза, проявляющийся пигментацией и лихенификацией в перианальной и крестцовой областях, однако у таких пациентов нередко имеются также очаги лихеноидного или бифазного амилоидоза в других зонах. При лихеноидном амилоидозе описаны и буллёзные элементы, хотя они чаще ассоциированы с системным амилоидозом.
Макулярный и/или лихеноидный амилоидоз также описан в ассоциации с аутоиммунными заболеваниями соединительной ткани, такими как системная склеродермия, системная красная волчанка и дерматомиозит, а также с первичным билиарным циррозом. У одного пациента было отмечено отсутствие очагов лихеноидного амилоидоза в зонах с более высокой температурой кожи, например по ходу крупных поверхностных вен, что позволяет предположить, что образование амилоидных фибрилл in vivo может быть температурозависимым процессом.
Семейные синдромы первичного кожного амилоидоза
Был выявлен ряд генетических заболеваний, предрасполагающих к развитию первичного локализованного кожного амилоидоза (PLCA).
Амилоидоз, ассоциированный с рецептором IL-31 (PLCA1 и PLCA2)
У нескольких семей с первичным локализованным кожным амилоидозом были обнаружены патогенные варианты в генах, кодирующих либо β-субъединицу рецептора онкостатина M (OSMR; PLCA1), либо α-субъединицу рецептора интерлейкина-31 (IL-31RA; PLCA2). Однако белковый продукт OSMR служит β-субъединицей не одного, а двух рецепторов — как OSMR, так и IL-31R; эта субъединица затем соединяется с IL-31RA, образуя рецептор IL-31. Следует отметить, что IL-31 играет важную роль при зудящих заболеваниях кожи, например при дерматитах и узловатом пруриго, а немолизумаб — антитело к IL-31RA — уменьшал зуд у пациентов с атопическим дерматитом. В этих семьях с первичным локализованным кожным амилоидозом на сигнальный путь IL-31, вероятно, также влияет моноцитарный хемоаттрактантный белок 1 (MCP-1).
Дисхромический амилоидоз [1] (PLCA3)
Дисхромический амилоидоз, также называемый первичным локализованным кожным амилоидозом 3-го типа (PLCA3), представляет собой необычный вариант, при котором каплевидная лейкодерма накладывается на фон гиперпигментации и сочетается с характерными проявлениями макулярного и лихеноидного амилоидоза. У пациентов с дисхромическим амилоидозом были выявлены гомозиготные и компаунд-гетерозиготные мутации в гене GPNMB, кодирующем гликопротеин nonmetastatic melanoma protein b.
Синдром Сиппла, или множественная эндокринная неоплазия (MEN) типа 2A
Синдром Сиппла — это аутосомно-доминантное заболевание, для которого характерна триада: медуллярный рак щитовидной железы, феохромоцитомы и гиперпаратиреоз. Повышение уровня кальцитонина в сыворотке отражает гиперплазию или карциному парафолликулярных C-клеток щитовидной железы, продуцирующих кальцитонин. Для множественной эндокринной неоплазии типа 2A характерны зудящие участки гиперпигментации в верхней части спины. Эти поражения описывались как notalgia paresthetica, макулярный амилоидоз и лихеноидный амилоидоз. Необычной особенностью является ранний возраст начала, часто до 10 лет. Распознавание этого кожного признака системного заболевания может привести к своевременному выполнению профилактической тиреоидэктомии после установления диагноза. В исследовании 10 семей с MEN 2A частота лихеноидного амилоидоза составила 36%; более того, у всех пациентов с кожным амилоидозом был выявлен определённый патогенный вариант в кодоне 634 протоонкогена RET.
Х-сцепленное ретикулярное пигментное нарушение (амилоидоз Партингтона)
Partington и соавт. описали семью с Х-сцепленным заболеванием, при котором у взрослых женщин отмечались линейные полосы гиперпигментации, а у мужчин наблюдалась ретикулярная, пятнистая коричневая пигментация кожи; при биопсии в дерме выявлялись отложения амилоида, причём у взрослых, но не у детей. У женщин поражение ограничивалось кожей, тогда как у мужчин также наблюдались рецидивирующие респираторные инфекции, дистрофия роговицы и светобоязнь. Как и при макулярном амилоидозе, амилоидные отложения дают положительную реакцию на кератин и отсутствуют в других тканях. У представителей нескольких семей были выявлены патогенные варианты в гене POLA1 на хромосоме Xp22, кодирующем каталитическую субъединицу ДНК-полимеразы α1. Это сопровождалось активацией провоспалительных генов, например NF-κB, и усилением интерферонового профиля I типа.
Другие ассоциированные заболевания
Первичный кожный амилоидоз также описан в ассоциации с врождённой пахионихией, врождённым дискератозом и семейной ладонно-подошвенной кератодермией. В местах инъекций некоторых лекарственных препаратов, например инсулина, также могут развиваться кожные амилоидные отложения.
Узловой амилоидоз
Узловой амилоидоз встречается редко и проявляется одиночными или множественными “восковидными” узлами либо инфильтрированными бляшками, чаще всего на туловище или конечностях. В составе амилоидных отложений выявлены γ-лёгкие цепи иммуноглобулинов, а также β2-микроглобулин; предполагается, что оба компонента продуцируются расположенными поблизости плазматическими клетками.
У ряда пациентов с узловым амилоидозом кожные плазматические клетки демонстрировали моноклональную рестрикцию лёгких цепей иммуноглобулинов, установленную с помощью иммуногистохимического исследования или гибридизации in situ; при исследовании костного мозга у одного пациента признаков клональной плазмоклеточной пролиферации выявлено не было. Следует отметить, что возможна ассоциация между синдромом Шегрена и узловым амилоидозом кожи или лёгких. Было высказано предположение, что по крайней мере часть случаев кожного узлового амилоидоза может представлять собой вариант первичного кожного лимфопролиферативного заболевания краевой зоны, то есть лимфомы, с необычными гистопатологическими признаками, но столь же благоприятным прогнозом.
Имеются сообщения о прогрессировании узлового варианта с развитием системного поражения, в связи с чем целесообразно длительное динамическое наблюдение. Однако более поздние данные показывают, что этот риск составляет около 7%. Хотя у 40% пациентов в этом длительном исследовании при первичном обследовании выявлялась парапротеинемия, гаммапатия оставалась стабильной в течение периода наблюдения, а прогрессирование до системного поражения было отмечено лишь у 1 из 15 пациентов — через 23 года после первичного обращения.
В аналогичном исследовании 16 пациентов с узловым амилоидозом распределение по полу было почти одинаковым, при этом отмечалась тенденция к акральной локализации высыпаний. Один пациент впоследствии умер от системного амилоидоза.
Диагностика
Диагноз первичного кожного амилоидоза основывается на клинической картине и гистологическом выявлении отложений амилоида в коже.
Патоморфология
Окраски, обнаруживающие амилоид:
- Гомогенные, гиалиноподобные, эозинофильные отложения в срезах, окрашенных гематоксилином и эозином
- Метахромазия при окраске кристаллическим фиолетовым (генцианвиолет)
- Положительное окрашивание щелочным конго красным
- Яблочно-зелёное двойное лучепреломление в поляризованном свете после окрашивания конго красным; также характерна двуцветность (от желто-зеленого до сине-зеленого)
- Яркое свечение при ультрафиолетовой флуоресцентной микроскопии после окрашивания тиофлавином T
- Окрашивание антителами к амилоидному P-компоненту
- Окрашивание антителами, направленными против специфических белков-предшественников, например кератина
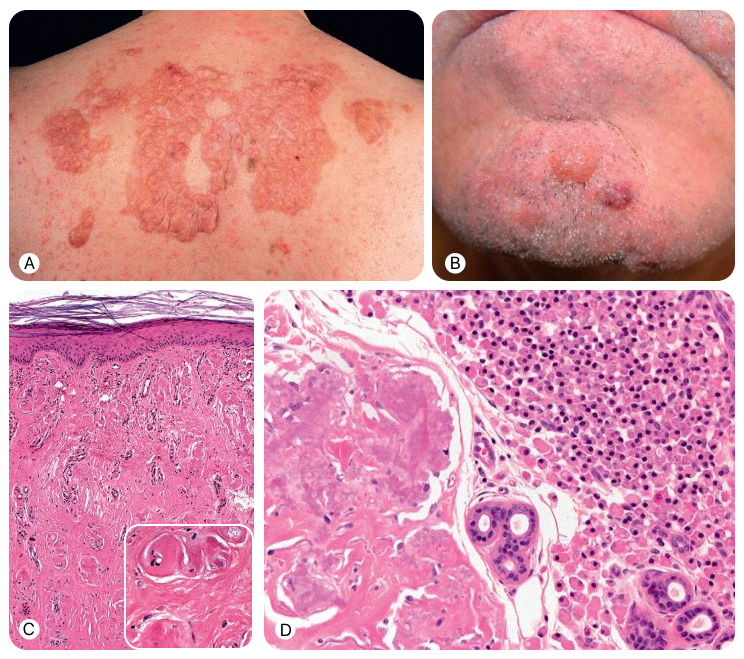
Макулярный и лихеноидный амилоидоз
- Амилоидные отложения ограничены верхними отделами дермы, преимущественно сосочковым слоем дермы.
- При лихеноидном амилоидозе эти отложения могут расширять дермальные сосочки и латерально смещать удлинённые эпидермальные выросты.
- В покрывающем эпидермисе часто выявляются признаки хронической травматизации, например акантоз и компактный ортогиперкератоз.
- При обеих формах могут обнаруживаться меланофаги и скудный периваскулярный лимфогистиоцитарный инфильтрат.
- Хотя антитела к кератинам, например MNF 116, могут быть полезны для подтверждения кератинового происхождения амилоидных отложений при этих двух формах первичного кожного амилоидоза, необходимость в них возникает редко.
Узловой амилоидоз
- Дерма, подкожная жировая клетчатка и стенки кровеносных сосудов диффузно инфильтрированы амилоидом.
- Периваскулярный инфильтрат из плазматических клеток обнаруживается почти всегда, однако в большинстве случаев он не выражен.
- эти плазматические клетки обычно являются моноклональными, и в последнее время было высказано предположение, что ряд случаев первичного кожного узлового амилоидоза («узловой амилоидомы») может представлять собой своеобразный вариант кожного лимфопролиферативного заболевания краевой зоны, то есть лимфомы.
- Иммуногистохимическое выявление отложения лёгких цепей может способствовать диагностике узлового амилоидоза.
Дифференциальная диагностика
Парестетическая ноталгия
Существует значительное клиническое сходство между макулярным амилоидозом и парестетической ноталгией. Последняя также локализуется в верхней части спины и может сопровождаться рябящей гиперпигментацией. Гистологически на ранних стадиях парестетической ноталгии выявляются рассеянные некротизированные кератиноциты и немногочисленные меланофаги, однако амилоидные отложения отсутствуют. Тем не менее на более поздних этапах, даже в пределах того же очага, такие отложения могут появляться.
Когда макулярный амилоидоз носит более диффузный или сливной характер, его необходимо дифференцировать с поствоспалительной гиперпигментацией. В дифференциальный диагноз также могут входить отрубевидный лишай, атрофический красный плоский лишай, стойкая дисхромическая эритема (ashy dermatosis) и лекарственно-индуцированная пигментация.
Лихеноидный амилоидоз
- Ограниченный нейродермит
- Гипертрофический красный плоский лишай.
Оба состояния характеризуются хроническими зудящими бляшками, часто локализующимися на голенях, и гистологически проявляются гиперкератозом, акантозом и слабо- или умеренно выраженным лимфогистиоцитарным воспалительным инфильтратом, однако без амилоидных отложений. При ограниченном нейродермите обычно выражена лихенификация, тогда как очаги гипертрофического красного плоского лишая могут иметь фиолетовый оттенок и при гистологическом исследовании сопровождаться лихеноидным инфильтратом с вакуольной дегенерацией.
В клинический дифференциальный диагноз также могут входить папулёзный муциноз, претибиальная микседема, узловатое пруриго, пемфигоидное узловатое пруриго, lichen ruber moniliformis и зудящая форма буллёзного эпидермолиза.
Узловой амилоидоз
Узловой амилоидоз может напоминать кожные проявления первичного системного амилоидоза, поэтому последний необходимо исключать.
В клинический дифференциальный диагноз также входят другие узловые поражения кожи, например кожная лимфоидная гиперплазия, саркоидоз и коллоидные миллиумы; все они имеют достаточно характерные гистопатологические признаки и потому обычно легко различимы.
[1] Kurian SS, Rai R, Madhukar ST. Amyloidosis cutis dyschromica. Indian Dermatol Online J 2013;4:344-6. PDF