Синонимы: lipoid proteinosis, Urbach - Wiethe disease, болезнь Урбаха-Вите
Главное
- Редкое аутосомно-рецессивное заболевание накопления, обусловленное мутациями в гене ECM1, кодирующем внеклеточный матриксный белок 1
- Гиалиноподобный материал откладывается в различных органах, включая кожу, слизистую оболочку полости рта, гортань и головной мозг
- Папулы и узелки преимущественно локализуются на лице; там же часто наблюдаются оспенноподобные рубцы. Диффузное восковидное утолщение кожи может сочетаться с веррукозными изменениями, особенно в области локтей, коленей и кистей
Историческая справка
- Липоидный протеиноз впервые был описан Siebenmann в 1908 году.
- Спустя двадцать лет это заболевание было выделено как самостоятельная нозологическая единица двумя венскими врачами — дерматологом Erich Urbach и оториноларингологом Camillo Wiethe.
Эпидемиология
Большинство пациентов имеют европейское происхождение, включая потомков голландских переселенцев, обосновавшихся в Южной Африке в середине XVII века. У африканеров не только отмечается наибольшая частота этого генетического заболевания, но и выявляется founder effect, то есть наличие общей мутации в популяции.
Патогенез
В 2002 году было показано, что липоидный протеиноз обусловлен мутациями с утратой функции в гене ECM1, кодирующем внеклеточный матриксный белок 1 (extracellular matrix protein 1/ECM1).
ECM1 представляет собой секретируемый гликопротеин, который может выступать как отрицательный регулятор энхондрального остеогенеза, промотор ангиогенеза и ингибитор матриксной металлопротеиназы 9. Кроме того, ECM1a (см. ниже) обнаруживается в зоне базальной мембраны и способен усиливать связывание коллагена IV с ламинином 332. Он также взаимодействует с компонентами внеклеточного матрикса, такими как фибронектин и гликозаминогликаны (например, гиалуроновая кислота, хондроитина сульфат).
У ECM1 имеются три основных изоформы:
- ECM1a — белок из 540 аминокислот, кодируемый геном из 10 экзонов;
- ECM1b — белок из 415 аминокислот, возникающий при отсутствии exon 7;
- ECM1c — белок из 559 аминокислот вследствие наличия дополнительного exon 5a в intron 5.
У пациентов с мутациями в 7-м экзоне сохраняется экспрессия изоформы ECM1b, и заболевание у них обычно протекает легче по сравнению с пациентами с мутациями в 6-м экзоне, у которых течение, как правило, более тяжелое и снижена экспрессия всех трех изоформ. Большинство патогенных мутаций локализуется в 6-м или 7-м экзоне; реже — в 2-м, 4-м или 9-м. Степень дермального иммунофлюоресцентного окрашивания с использованием anti-ECM1 антител позволяет косвенно судить о локализации мутации: сниженное окрашивание характерно для мутаций в 7-м экзоне, а отсутствие окрашивания — для мутаций в 6-м экзоне.
Клиническая картина
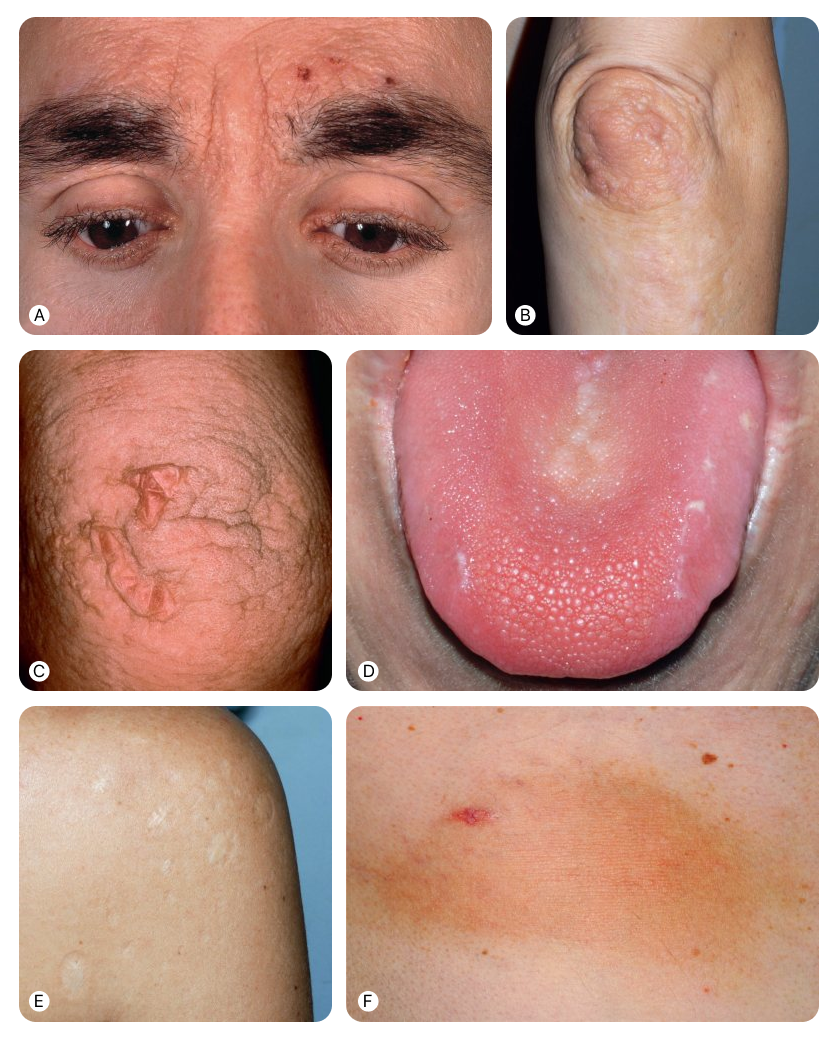
Первым клиническим признаком липоидного протеиноза часто бывает слабый крик или охриплость у новорожденных, обусловленные инфильтрацией слизистой оболочки гортани. Охриплость сохраняется на протяжении всей жизни. Кожные проявления обычно возникают в течение первых двух лет жизни и проходят две частично перекрывающиеся стадии.
На первой стадии в полости рта, а также на коже лица и конечностей возникают везикулы и геморрагические корки, нередко после травматизации. Кожные элементы могут разрешаться с образованием рубцов, в том числе по типу «ледоруба/ice-pick».
На второй стадии происходит нарастание гиалиновых отложений в дерме. Кожа становится диффузно утолщенной, восковидной и приобретает желтоватый оттенок. На лице, включая края век, а также в подмышечных впадинах и на мошонке появляются папулы, бляшки и узлы. На разгибательных поверхностях, особенно в области локтей, коленей и кистей, могут формироваться веррукозные очаги. Описаны также генерализованное шелушение и алопеция бровей и ресниц. Из-за инфильтрации век возможно образование язв роговицы.
Помимо поражения языка (диффузная инфильтрация), уздечки языка (ограничение подвижности языка) и ротоглотки, вследствие окклюзии слюнного протока может возникать рецидивирующий паротит. К стоматологическим аномалиям относятся гиперплазия или аплазия верхних резцов, премоляров либо моляров; обычно пациенты теряют зубы в раннем возрасте.
Неврологические проявления встречаются часто и включают поведенческие и обучающие трудности, судороги и, реже, внутримозговое кровоизлияние. Патогномоничным рентгенологическим признаком является двусторонняя внутричерепная серповидная кальцификация в области миндалевидных тел. В более поздних наблюдениях также описана повышенная частота кист почек.
В целом заболевание имеет стабильное или медленно прогрессирующее течение и совместимо с нормальной продолжительностью жизни, за исключением риска респираторной обструкции в младенческом возрасте. Может отмечаться повышение СОЭ вследствие увеличения уровня α- или γ-глобулинов.
Патоморфология
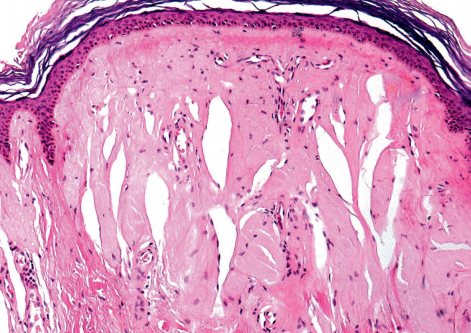
Основные клинические проявления связаны с отложением аморфного или пластинчатого материала вокруг кровеносных сосудов и в соединительной ткани. Аморфные отложения состоят преимущественно из неколлагеновых белков, тогда как концентрические слои материала, напоминающего базальную мембрану, содержат коллаген II и IV, а также ламинин. Кроме того, отложения PAS-позитивны и диастазорезистентны, что указывает на наличие нейтральных гликозамингликанов (ГАГ).
В срезах, окрашенных гематоксилином и эозином, в ранних очагах выявляются розовые гиалиноподобные отложения вокруг капилляров в сосочковом слое дермы и по периферии эккринных потовых желез. Более поздние очаги характеризуются гиперкератозом, иногда папилломатозом, и утолщенной дермой, где пучки розовых гиалиновых отложений располагаются диффузно; эти пучки нередко ориентированы перпендикулярно дермоэпидермальному соединению. В нижних отделах дермы могут обнаруживаться более мелкие рассеянные гиалиновые отложения. Гиалин также окружает волосяные фолликулы, сальные железы, мышцы, поднимающие волос, а также эккринные железы.
Помимо коллагена IV, ламинина и нейтральных ГАГ, в дерме присутствует гиалуроновая кислота, что подтверждается окраской альцианом синим. Окраски на жиры (например, Судан III) дают непостоянные результаты при фиксации ткани в формалине, а в редких случаях отложения окрашиваются конго красным.
Гистопатологические особенности везикулезной первой фазы описаны лишь в немногих работах и варьируют от внутриэпидермального акантолиза до малоклеточных субэпидермальных пузырей.
Дифференциальная диагностика
Клинический дифференциальный диагноз включает эритропоэтическую протопорфирию (ЭПП), амилоидоз, папулезный муциноз, коллоидные миллиумы и нелангергансоклеточные гистиоцитозы. На ранней стадии может рассматриваться световая оспа. У младенцев необходимо учитывать возможность синдром гиалинового фиброматоза (ювенильный гиалиновый фиброматоз и инфантильный системный гиалиноз), обусловленного мутациями в гене, кодирующем белок морфогенеза капилляров 2.
Гистологически дифференциальный диагноз включает ЭПП и коллоидные миллиумы. В кожных очагах при ЭПП, коллоидных миллиумах и амилоидозе гиалинизация выражена слабее, более очаговая и поверхностная; эккринные железы вовлекаются редко. При коллоидных миллиумах отложения не имеют столь выраженного периваскулярного рисунка, как при липоидном протеинозе.
Лечение
В настоящее время излечивающего или доказанно эффективного лечения липоидного протеиноза не существует.
Применялись пероральные, интраочаговые (в том числе субмукозные) и топические кортикостероиды с некоторым положительным эффектом, а также увлажняющие средства. Успешно выполнялись пластические операции, дермабразия и лазерная шлифовка. Имеются ограниченные сообщения об использовании системных ретиноидов (например, Ацитретин 0,5 мг/кг/сут в течение 6 месяцев), D-пеннициламин и перорального диметил сульфоксид (DMSO). 1